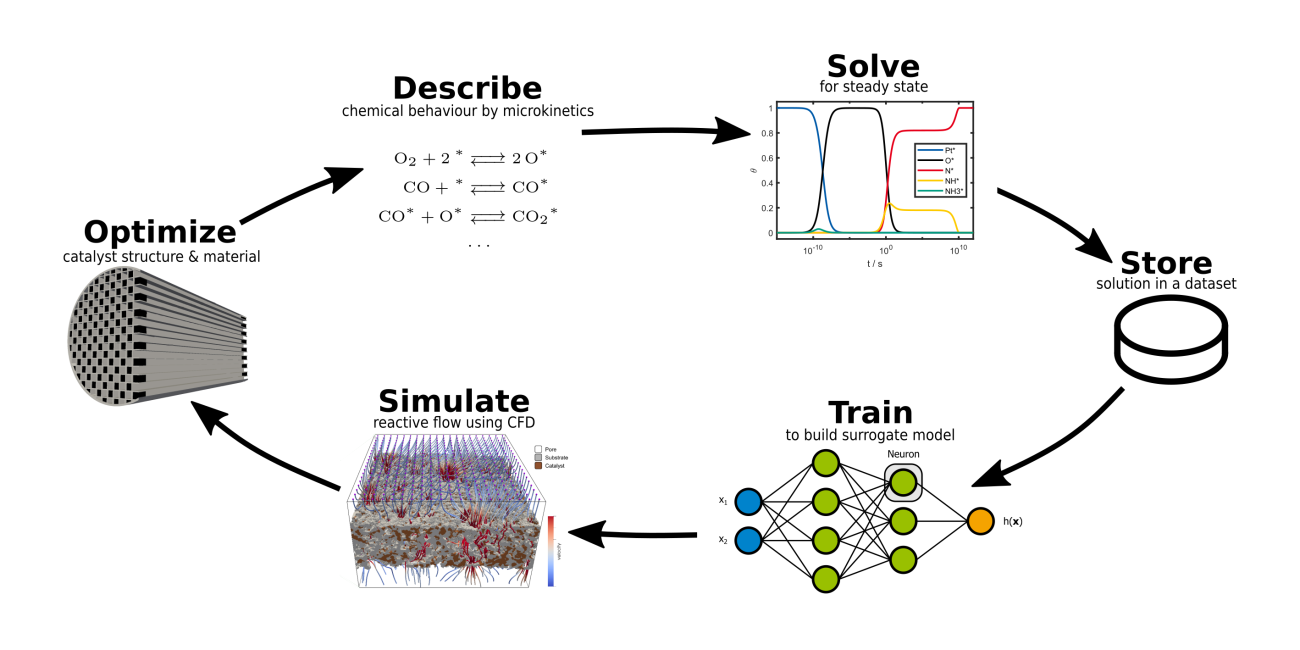
Reaction engineering aims to optimize transport processes in order to achieve high conversion and selectivity and ensure safe operation. Computational fluid dynamics provides detailed information about these transport processes and is therefore a valuable tool in reactor simulation when chemical reactions are incorporated. While our mechanistic understanding of chemical reactions is ever-growing, using this knowledge in full-scale reactor simulations turns out to be challenging. Coupling microscopic length and time scales of chemical processes with the macroscopic behaviour of the reactor leads to very demanding computations. Therefore, systematic investigations using our detailed understanding are expensive or even impossible.
Our goal is to speed up reactor simulations by developing models that predict the rate of chemical reactions precisely while maintaining feasible simulation times. As we blend physico-chemical understanding with Machine Learning to enhance modelling and simulation, our research can be referred to as Scientific Machine Learning. Examples of our daily work include:
- building a database of accurate steady state solutions of complex reaction networks,
- developing suitable data transformation techniques and
- training artificial Neural Networks to perform regression by supervised learning.
However, we cover different model types as well: Inspired by the possibility of intelligent data sampling we use Random Forests, cooperate with Uni Stuttgart to deploy Kernel Methods and have had experience with spline interpolation for years.
For our current project ML MORE, we model CO oxidation kinetics to improve simulation capabilities of the engine exhaust gas flow through the complex porous structure of diesel particulate filters under a vast variety of conditions.
References
- Döppel, F. A., & Votsmeier, M. (2022). Efficient machine learning based surrogate models for surface kinetics by approximating the rates of the rate-determining steps.
Chemical Engineering Science, 262, 117964.
Click here for the freely available preprint version - Döppel, F. A., & Votsmeier, M. (2023). Efficient Neural Network Models of Chemical Kinetics Using a Latent asinh Rate Transformation.
Reaction Chemistry & Engineering, Accepted Manuscript
