

Coronavirus and Disinfectant
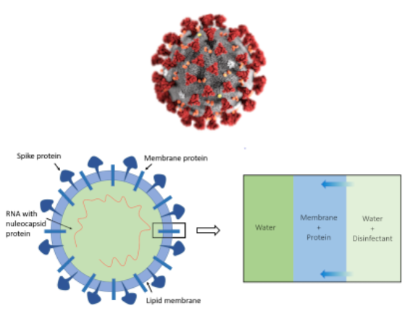
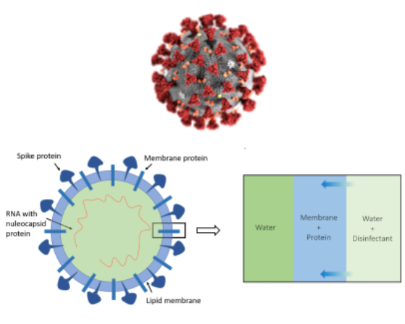
The project investigates by molecular dynamics simulations the action of common disinfectants such as ethanol or isopropanol on the integrity of the external membrane of the coronavirus.
For further information go to: ccteamcov.blogspot
Disclaimer: We are using this channel instead of peer-reviewed publishing because of speed. We want to make results available to practitioners in time for them to be useful in the current crisis. Therefore, results may be preliminary, not quite converged or inaccurate. Still they might be useful, and they are the best we can do at the time. Use our results with care.
Mobility of polymer melts in confinement
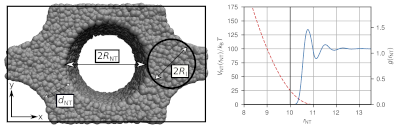
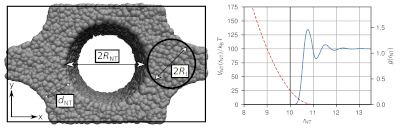
The development of polymer nanocomposites is of great scientific interest and especially carbon nanotubes show promising properties, such as electrical and thermal conductivity, impact strength or viscoelasticity. Material properties strongly depend on the alignment of nanoparticles and polymers. We study entangled polymers in confinement in order to get an insight into the mobility behavior of the polymers. We use Dissipative Partivle Dynamics (DPD) as our coarse-grained simulation method together with a slip-springs model, which has been developed in our group. This model has shown to reproduce entangled dynamics of polymer chains. For soft-core methods, like DPD, polymer dynamics are generally greatly accelerated. The confinement in this work consists of nanotubes, arranged in a hexagonal array. These nanotubes are modeled as excluded volume through a repulsive potential and we analyze structural and dynamic properties of the polymers. Our model yields the potential to be further refined by a more structured nanotube model and a more detailed analysis of the chain movement. Moreover, irregular arrays or the dispersion of nanotubes under a flow of entangled polymers could then be studied in future projects.
For further information please contact Simon Alberti
Investigating momentum conservation in hybrid Particle-Field models
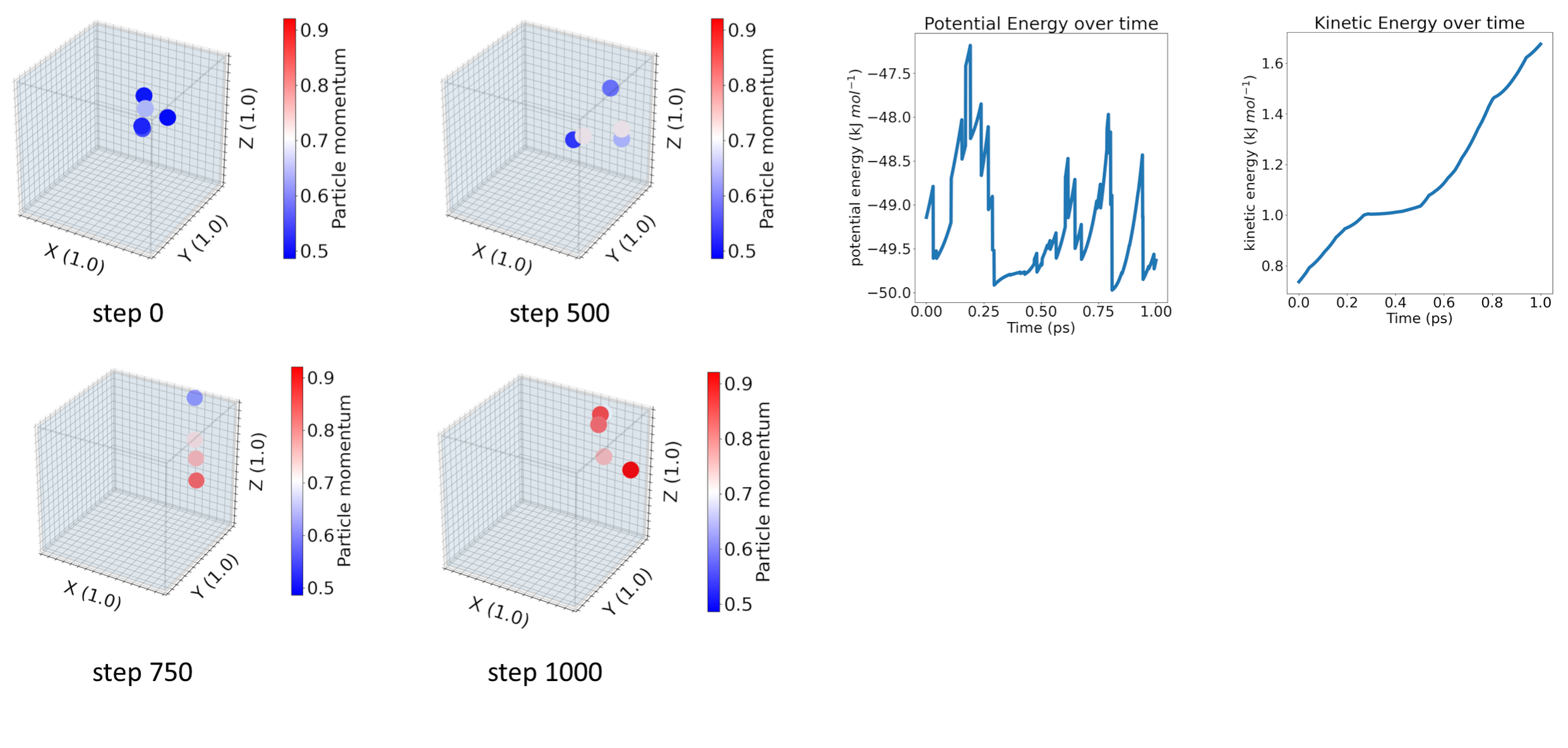
Accurate prediction of viscosities and other rheological properties of polymer melts is an important step towards energy-efficient plastic processing and recycling. For this purpose we use hybrid Particle-Field (hPF) models, a simulation method that combines particle models with density fields. In hPF models intermolecular interactions are derived from an external potential, generated based on the density field. This approach has been shown to be more efficient than the intermolecular pairwise interactions used in standard atomistic or coarse-grain molecular dynamics (MD) simulations. However, while hPF models have been effectively used in a variety of static properties of polymer melts, biological systems, and nanoparticles, they are less successful when it comes to transport properties. This is because the external field breaks the Galilean invariance, making hPF models not strictly momentum conserving. In this project, our goal is to investigate the possibility of making hPF models momentum conserving at machine scale precision. For this purpose, we focus on studying the dynamics at single to few particle systems. This allows us to revisit the fundamental concepts of the hPF method, such as the calculation of the external potential and the placement of the density grid, and prove our methods analytically before studying more complex larger-scale systems.
For further information please contact Evangelia Charvati
Wetting behavior of underwater oil droplets on superhydrophilic surfaces
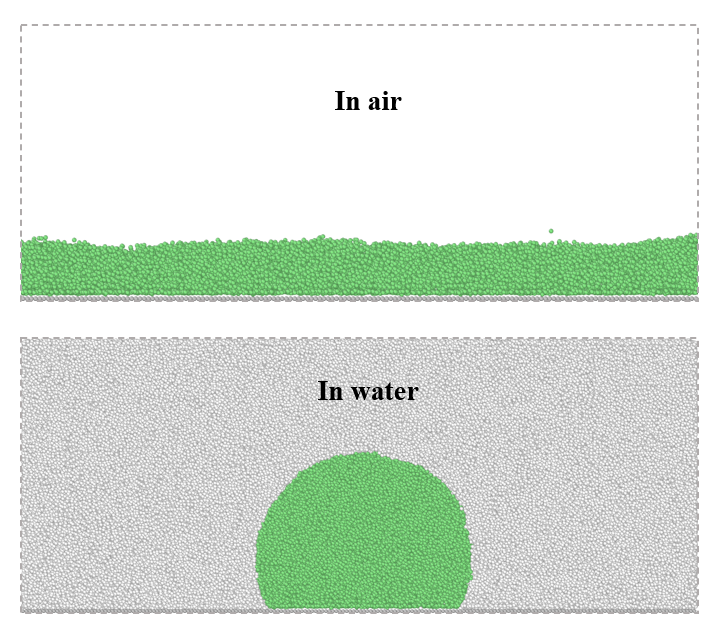
Achieving superoleophobic surfaces in air is more challenging than creating superhydrophobic surfaces as oils have a much lower surface tension. However, recent experimental studies have shown that a surface exhibiting superhydrophilicity in air can commonly show superoleophobicity when submerged in water. The underwater Cassie state, which is crucial for realizing underwater superoleophobicity, can be achieved by forming special micro/nano structures on hydrophilic surfaces or grafting hydrophilic polymer brushes onto them. In this project, we investigate the wetting behavior of underwater oil droplets on superhydrophilic surfaces, using many-body dissipative particle dynamics. The main aim of this project is to understand the relationship between the properties of the polymer brush layer and its underwater oil-repellent performance.
For further information please contact: Xinxin Deng
Designing Compatibilizers for Recycled Polymers Using DPD Simulations
Recycled polymer blends often consist of immiscible components, such as PET, PP, and PE, resulting in poor mechanical properties and limited processability. This project aims to develop a systematic approach for designing effective compatibilizers to enhance interfacial properties and improve the performance of these blends. By combining Hansen solubility parameters (HSP) and Dissipative Particle Dynamics (DPD) simulations, we analyze the thermodynamic behavior of polymer interfaces and quantify the impact of compatibilizers on interfacial tension. A key focus is the comparison of different compatibilizer architectures, such as graft and multiblock copolymers, to determine their efficiency in reducing interfacial tension and stabilizing polymer interfaces.
For further information please contact Kosar Khajeh
Multiscale Modes based on Molecular Roughness
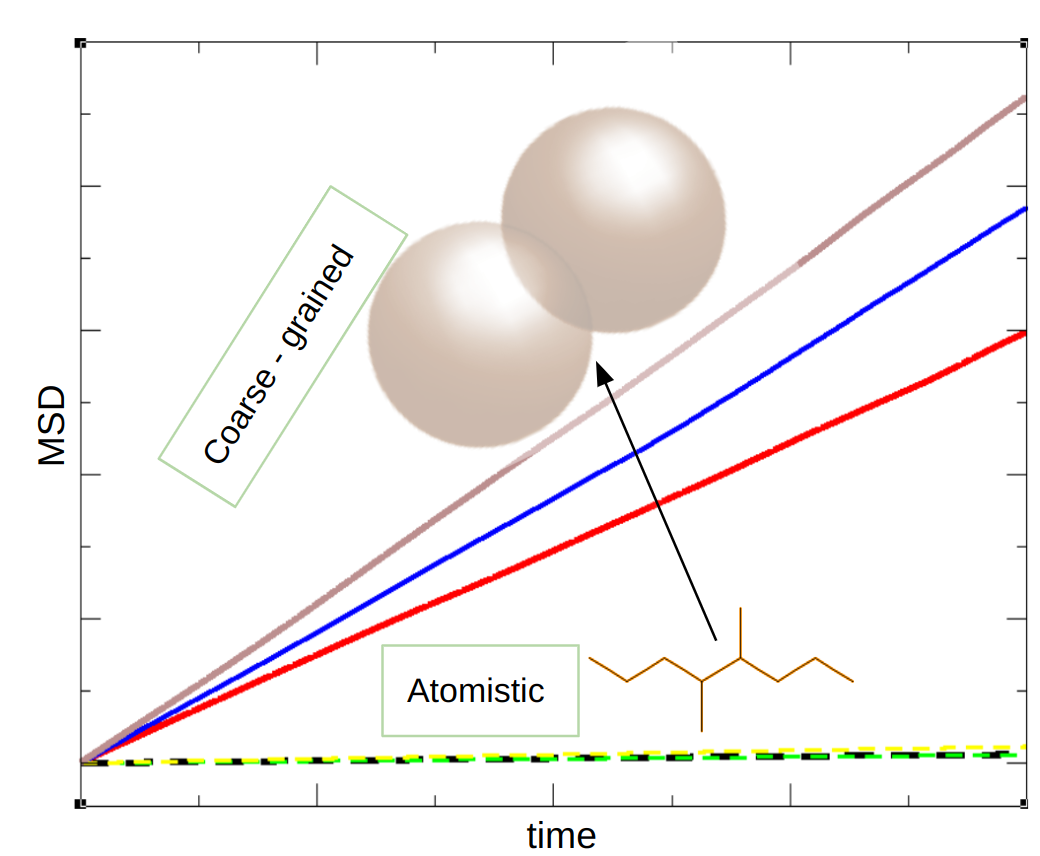
Different methods and techniques have been developed to predict the change in structural and dynamic properties upon changing the formulation of polymeric material. In recent years, the molecular simulation of materials relies increasingly on coarse‐grained models. These models are simpler than fully atomistic models and allow the simulation of larger systems for longer time scales. Coarse-grained models include the most important degrees of freedom in the computation that are necessary to study the dynamical properties of the system. Because of the reduction in degrees of freedom, the dynamics of the coarse-grained system gets more accelerated as compared to the all-atomistic system or their experimental values. Based on different considerations, there are several different ways that explain the increased molecular mobility upon coarse-graining. In this project, we will choose an entirely different scheme that is based on the “mechanical” consideration is known as the Rough-Mob method. This method is basically an approach to linking the increase of molecular mobility to the loss of molecular surface roughness as we move from an all-atomistic to a coarse-grained model.
For further information contact: Manisha
Multi-scale MD-FE coupled simulations of polymers
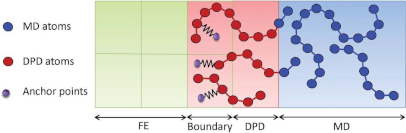
When simulating materials, one often encounters the situation that only in a small part of the material something “interesting” happens, but where the whole of the material needs to be described, as the less interesting regions influence directly or indirectly what is going on in the interesting region. An example is the failure of a polymer composite: When it breaks, complicated rearrangement of the polymer molecules take place near the crack tip. They are interesting from a fundamental and an application viewpoint. Away from the crack tip, the polymer is deformed under the load and this deformation will influence the processes at the crack tip. However, the deformation of the outer regions is not in itself interesting. Another example is the process of dry adhesion. For such cases, an algorithm is being developed by the Müller-Plathe group which allows us to conduct Molecular Dynamics – Finite Elements method (MD-FE) coupled Multi-scale Simulations.
For further information please contact Saeed Norouzi
Wetting behaviour of a droplet on nanostructured hydrophobic surfaces
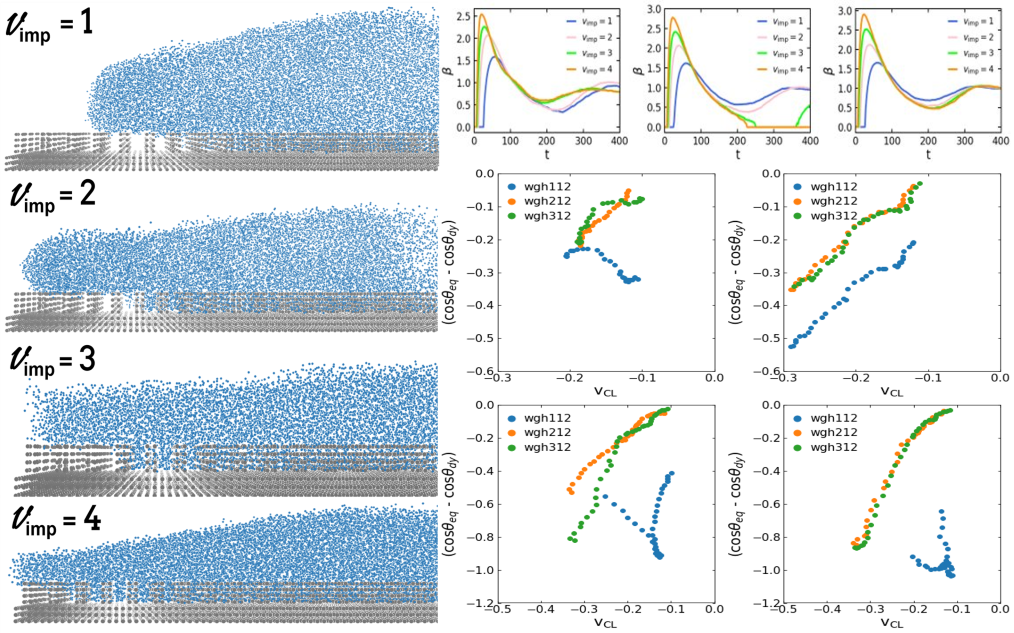
The static and dynamic wetting behaviour of a droplet on a hydrophobic rough surface is influenced by multiple parameters such as the surface topography, droplet properties, and kinematic parameters. Using Multi-Body Dissipative Particles Dynamics (MDPD), we study the static and dynamic wetting behaviour of a droplet on hydrophobic surfaces structured with square-shaped pillars. We calculate the maximum spreading factor and the dynamic contact angle. Within the framework of the Molecular Kinetic Theory (MKT), we also focus on understanding the dynamic wetting process by obtaining the effective friction coefficient of the contact line and comparing them depending on the selected surface topography and the impact velocity. The goal of the project is to understand the influence of the surface topography and impact velocity on the wetting behaviour of the droplet.
For more information please contact: Dejian Qiu
Multiscale Simulation of Plant Seed based Pectin
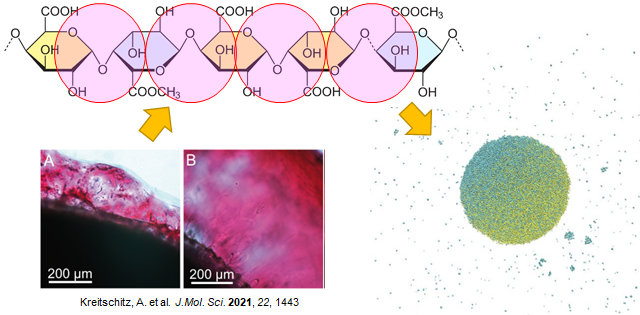
After hydration, some plant seed coats can produce the mucilage envelope. The adhesive property of this mucilage enables the seed to stick to the ground and dispersal over a long distance by sticking the seed to the bodies of animals. The main components of the mucilage are pectin, which is characterized by a strong capacity for hydration and for forming a gel-like envelope after wetting. From the experimental result, increasing the drying time of the mucilage seed decreases the contact angle and increases the adhesive force. This is because losing water makes mucilage becomes harder and stiffer. This project is about developing a coarse-grained model based on the chemical pectin oligosaccharide to reproduce mechanical properties. The effect of desiccation time on the adhesive force of pectin will be examined by multi-scale simulation. In addition, the adhesive property of pectin droplets with different compositions (pure pectin and pectin mixed with cellulose) will also be investigated.
For further information please contact: Kamonthira Wichai